Research
Our primary interest lies in understanding how genes are regulated and, more specifically, finding out new modes of gene regulatory mechanisms and developing methods to inspect how the molecules are wired to each other. In the meantime, we are focusing on post-transcriptional regulations by the RNA binding proteins using high-throughput biochemistry and high-throughput nucleic acid sequencing. The transcriptome- and proteome-wide views by the high-throughput methods allow us to step closer to the rules of nature with less bias towards our current knowledge.
Optimal Design Strategies for mRNA Vaccines
mRNA vaccines use untranslated regions (UTRs) and coding sequences (CDS) similar to cellular mRNAs. A single protein sequence can have over 10600 possible mRNA sequences due to codon redundancy, and the diversity in UTRs is even greater. Unlike cellular mRNAs, mRNA vaccines are not replicated inside the cell, so optimizing their stability is crucial for efficacy.

We are developing methods to rapidly design optimal mRNA sequences by considering biological factors that influence RNA stability. RNA secondary structure significantly affects stability, and our algorithms identify unstable features to guide vaccine design. Since RNA exists as an ensemble of multiple structures rather than a single conformation, it is essential to consider the thermodynamic probabilities of all possible structures during design.
We also focus on preserving cis-regulatory elements in UTRs for RNA stability while optimizing the overall sequence. To meet CEPI’s 100 Days Mission, we incorporate parallel cloud computing and design strategies that minimize DNA synthesis and assembly times.
Our optimization methods and algorithms are implemented in an open-source tool named VaxPress, with ongoing improvements to enhance stability and speed of mRNA therapeutic development.
Single-Molecule RNA Analysis in a Sub-Nucleotide Resolution
RNA molecules, despite originating from a common genome, exhibit remarkable diversity in their modifications and life cycles. This diversity arises from a range of processes, including polyadenylation, splicing, base modifications, capping, folding, subcellular localization, and interactions with numerous RNA-binding proteins. To elucidate the intricate regulatory mechanisms that bridge genomic DNA and protein expression, it is crucial to analyze individual RNA molecules at the single-molecule level.
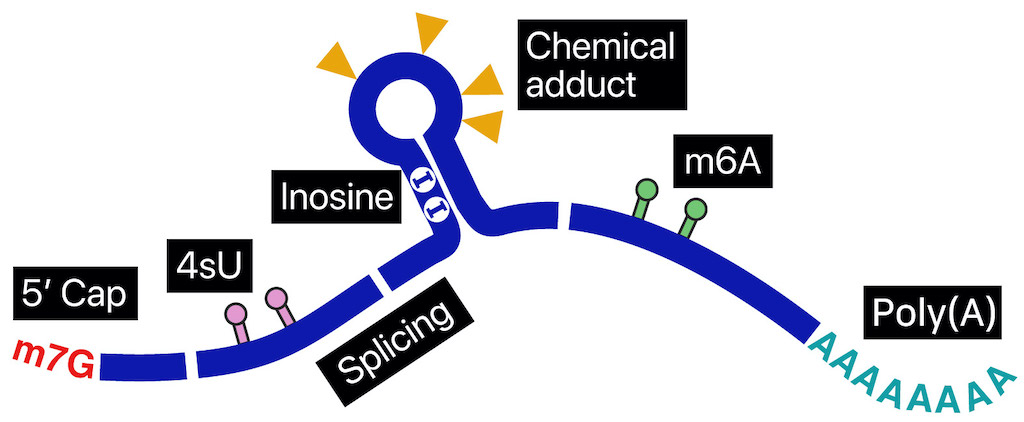
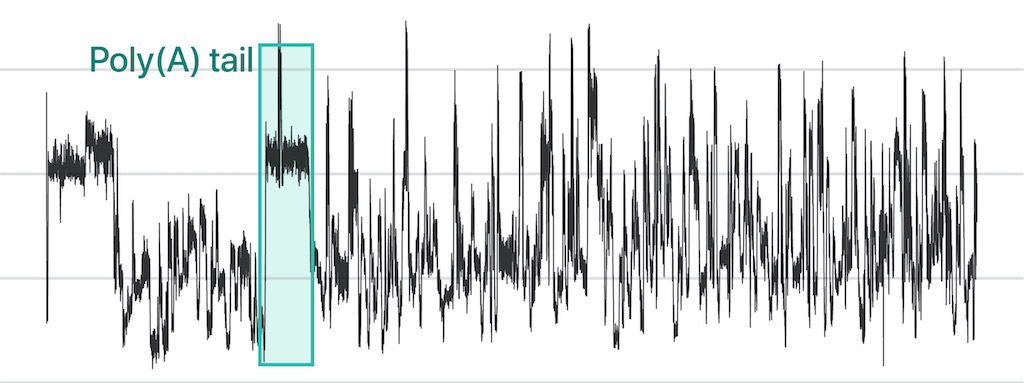
Nanopore direct sequencing has emerged as a transformative technology capable of revealing the complex connections within genetic circuitry. In nanopore direct RNA sequencing (DRS), nucleic acids traverse a nanoscale pore embedded in a bilayer membrane. As each RNA base passes through the nanopore, it induces a unique disturbance in the ionic current, enabling the detection of individual nucleotides. This technique offers unprecedented opportunities to examine sub-nucleotide-level details across full-length RNA molecules.
To leverage the potential of DRS for RNA biology, we are developing a comprehensive suite of biochemical methods and machine learning applications. These tools aim to enhance our ability to investigate previously unclear aspects of RNA regulatory mechanisms. By adapting direct RNA sequencing to the nuances of RNA biology, we seek to deepen our understanding of the complex processes that govern RNA function and regulation.